|
|

|

|
| e-Pub |
Section: New Results
Sequences
Random generation
The random generation of decomposable combinatorial structures, pioneered by P. Flajolet in the 80s, provides an elegant, yet powerful, framework to model and sample the objects which appear in computational biology. Random samples can then be used to assert the significance of a given observable when closed form formulae are difficult to obtain.
|
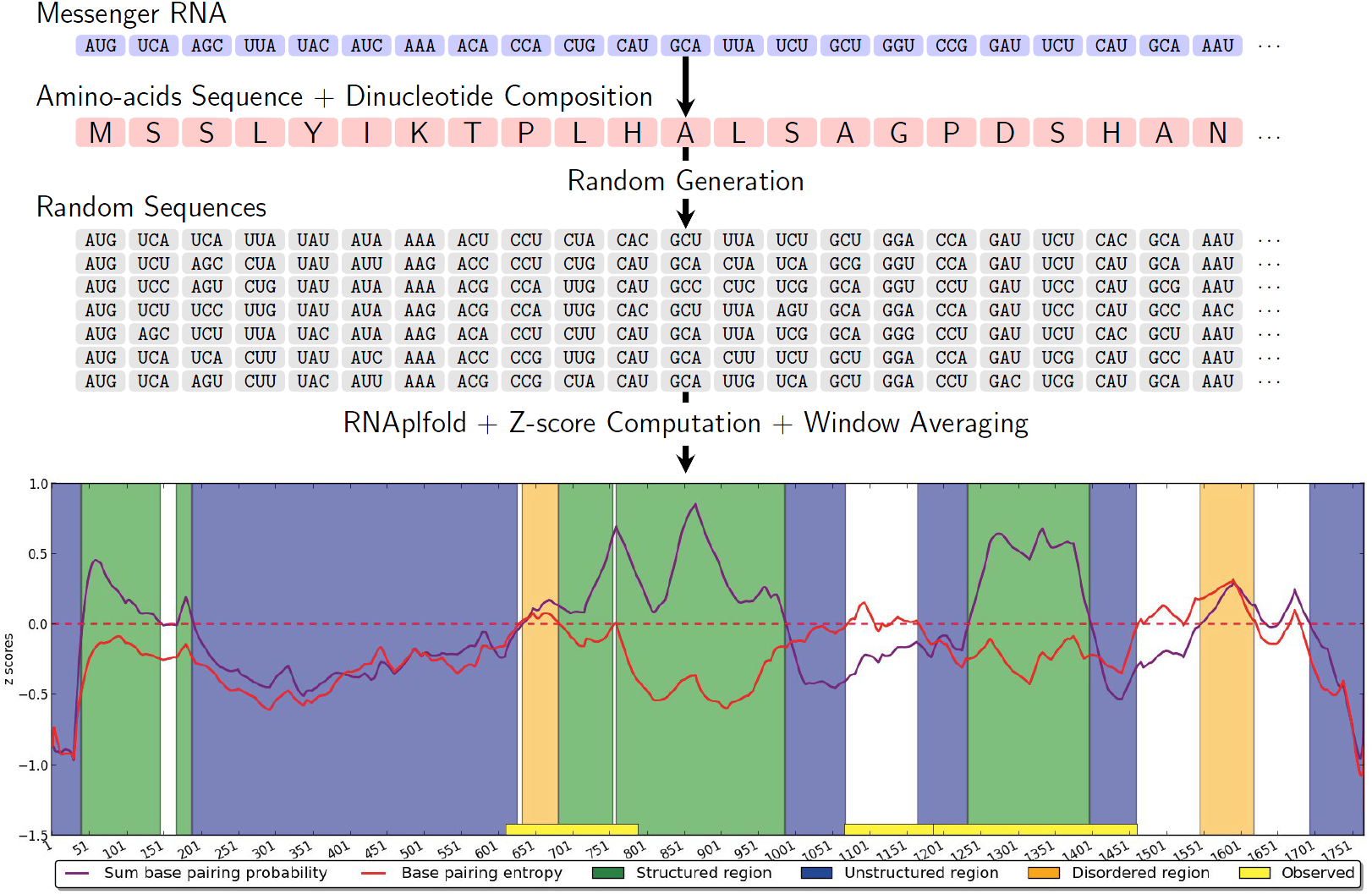
Messenger RNAs (mRNAs) encode proteins, but may also independently feature structured motifs which are crucial to recoding and alternative splicing mechanisms. In order to predict such motifs, the stability of smaller regions within a given mRNA must be compared to that of sequences generated with respect to a background model which, at the same time, preserves the encoded amino-acid sequence and the capacity of the overall sequence to form a stable fold (proxy-ed by the dinucleotide composition). Using multidimensional Boltzmann sampling, we have revisited the underlying – well-defined, yet never solved exactly – random generation problem, and provided the first unbiased and practical algorithm for the problem [27] . The algorithm, developed in collaboration with McGill and Université de Montréal (Canada), has linear time complexity as soon as a small tolerance (typically ) on the composition is allowed.
|
Some other biological objects, such as RNA secondary structures, naturally appear with probabilities which are poorly modeled by the uniform distribution. To better model such objects, Denise et al [3] have introduced the weighted distribution, and adapted classic random generation algorithms such that each object within a given combinatorial family can be generated with respect to it. However, the exponentially increasing probability ratio between the most and least probable object sometimes leads to a large degree of redundancy within generated sets . To work around this issue, and generate non-redundant sets of objects, we have proposed a sequential algorithm with deterministically avoids any previously generated word, without introducing any bias in the generation [17] .
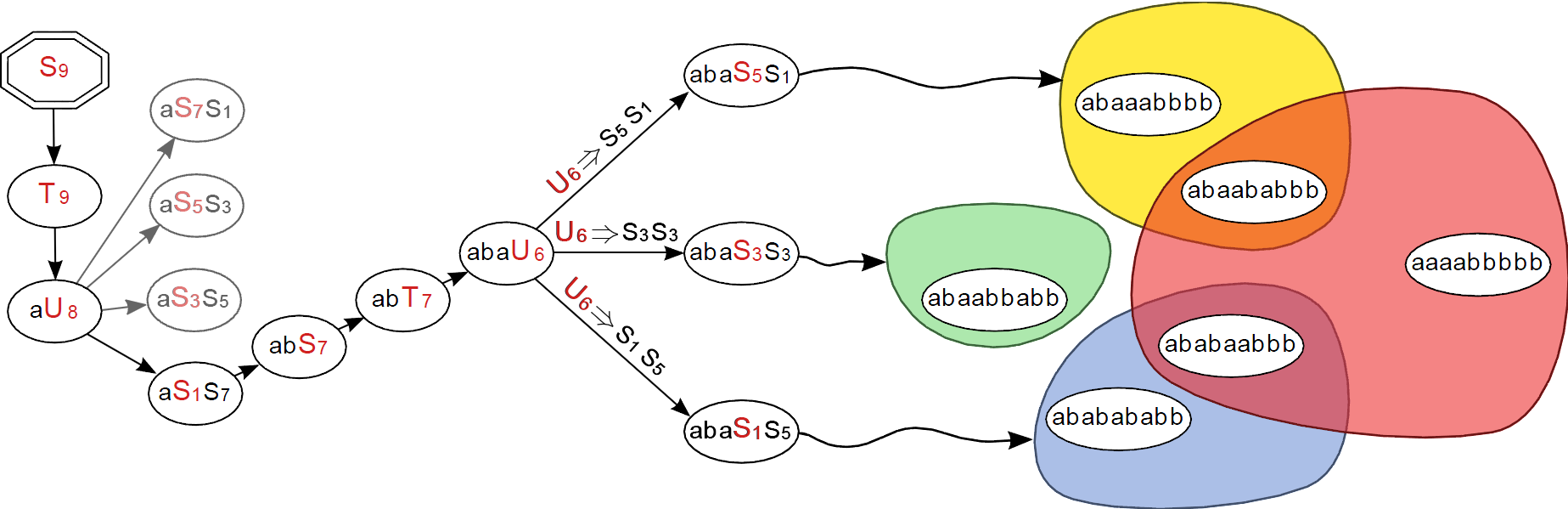
Besides, in collaboration with the Fortesse group at Lri , we developed a new divide and conquer algorithm for the random generation of words of regular languages, and we performed a complete benchmarking of all state-of-the-art methods dedicates to this problem [56] .
|

Next Generation Sequencing (NGS)
As a side-product of our previous collaborative studies with J. Waldispühl (McGill, Canada), focusing on sequence/structure relationship in RNA, we revisited the problem of detecting and correcting RNA sequences obtained using pyrrosequencing techniques. Indeed, ribosomal RNAs are often used to estimate the population diversity within a microbiome, and sequencing errors may lead to biased estimates. In this context, we investigated whether a complete knowledge of the RNA secondary structure could be exploited to detect and correct errors in NGS reads.
To that end, we introduced a probabilistic model, defined over all sequences at maximal distance from the input read and their respective folding. This model captures both the stability of the induced fold and its compatibility with a reference multiple sequence alignment. We designed a linear-time inside/outside algorithm to compute exactly the probability that a given position is mutated in the ensemble. Our initial implementation, presented at Recomb '13 [29] and published an extended version in Journal of Computational Biology [23] , revealed encouraging results, and we plan to combine it with a population diversity estimator to test its potential in a metagenomics context.
Combinatorics of motifs
An algorithm for pvalue computation has been proposed in [44] that takes into account a Hiddden Markov Model and an implementation, SufPref , has been realized (http://server2.lpm.org.ru/bio ).
Combinatorics of clumps have been extensively studied, leading to the definition of the so-called canonic clumps. It is shown in [28] that they contain the necessary information needed to calculate, approximate, and study probabilities of occurrences and asymptotics. This motivates the development of a clump automaton. It allows for a derivation of pvalues, decreasing the space and time complexity of the generating function approach or previous weighted automata.
Large deviations approximations are needed for very rare events, e.g. very small pvalues, as Gaussian approximations are known not to be applicable. In [21] , combinatorial properties of words allow to provide an explicit and tractable formula for the tail distribution with a low space and time complexity and a guaranteed tightness. Double strands counting problem is addressed where dependencies between a sequence and its complement plays a fundamental role. A large deviation result is also provided for a set of small sequences, with non-identical distributions. Possible applications are the search of cis-acting elements in regulatory sequences that may be known, for example from ChIP-chip or ChipSeq experiments, as being under a similar regulatory control. In a recent internship at Lix , F. Pirot detected a Chi-like motif in Archae genome.
In a collaboration with AlFarabi University, where M. Régnier acts as a foreign co-advisor), word statistics were used to identify mRNA targets for miRNAs involved in various cancers [8] , [9] .